


For isolated, free-flying nanoparticles or large molecules in many astrophysical environments, photon absorption is often the dominant excitation process. Prior to the absorption of an energetic photon, a molecule stays at the ground electronic state, which is S0 (the lowest singlet state) for neutrals and D0 (the lowest doublet state) for ions, containing very little vibrational energy. After photoabsorption occurs, the molecule is excited to an upper electronic state Sn for neutrals or Dn for ions (n > 1). The electronically excited molecule has 3 major competing decay channels to relax its energy: radiation, photoionization, and photodissociation. In this section we will focus on the radiation process and use PAHs as an example.
3.1. An Overview of the Photoexcitation and Emission Processes
Once the molecule is electronically excited, both radiative and radiationless transitions will occur between its vibrational modes, electronic and vibrational states (see Birks 1970 for detailed discussions on neutral molecules):
the molecule will undergo internal vibrational redistribution (IVR), an iso-energetic radiationless process between vibrational modes, which rapidly (~ 10-12-10-10 s) spreads the absorbed energy among its vibrational degrees of freedom;
the molecule will undergo
internal conversion (IC), a radiationless transition between
electronic states of the same multiplicity,
which very quickly (~ 10-12-10-8 s)
transfers the electronic energy to a highly vibrationally
excited state of the lower lying electronic state
(i.e., Si
 Sj for neutrals,
and Di
Dj for ions where i > j);
Sj for neutrals,
and Di
Dj for ions where i > j);
the molecule will also undergo
intersystem crossing (ISC),
a rapid radiationless transition
between electronic states of different multiplicity
(i.e., Si
Tj for neutrals, and Di
Qj for ions where i > j);
the molecule will undergo fluorescence,
a radiative electronic transition (~ 10-7 s)
between states of the same multiplicity,
which results in the emission of a visible photon
(i.e., Si
Sj for neutrals, and Di
Dj for ions where i > j;
for many molecules, this transition mainly takes place
between the first excited electronic state S1 or
D1
and the ground state S0 or D0, as a
consequence of rapid internal conversion which results in a practically
complete conversion of the initial excitation energy
to vibrationally excited levels of the S1 or
D1 and S0 or D0
electronic states);
the molecule will also undergo phosphorescence
with emission of a visible photon, a very slow (~ a few seconds)
radiative electronic transition between
states of different multiplicity (i.e., Ti
Sj for neutrals where i > j;
for ions, this transition [i.e. Qi
Dj] is not important
since internal conversion dominates over intersystem crossing
so that the molecule ends up in highly vibrationally excited
levels of the ground electronic state D0
[Leach 1987]);
finally, the molecule will undergo IR emission,
a radiative vibrational transition (~ 0.1 s)
between higher and lower vibrational states
of the same electronic state.
This transition mainly takes place in the ground electronic
state S0 or D0 because of rapid
internal conversion
and electronic fluorescence which ultimately drive the molecule
down to the ground electronic state with a high vibrational energy,
except for some neutral molecules (e.g. chrysene
C18H12)
a large fraction of this transition occurs in the first excited
triplet state T1 as a result of rapid intersystem
crossing. Note that in Figure 1 we only plot the
 v = 1 vibrational
transitions (where v is the vibrational quantum number)
given by the harmonic oscillator selection rule;
but overtone transitions with
v = 2,3,... are also
allowed when account is taken of anharmonicity.
We should stress that in astrophysical literature,
vibrational transitions are sometimes also called
"vibrational fluorescence" (e.g. see
Allamandola et al. 1989);
in this case, we shall call the fluorescence process
described above "electronic fluorescence".
v = 1 vibrational
transitions (where v is the vibrational quantum number)
given by the harmonic oscillator selection rule;
but overtone transitions with
v = 2,3,... are also
allowed when account is taken of anharmonicity.
We should stress that in astrophysical literature,
vibrational transitions are sometimes also called
"vibrational fluorescence" (e.g. see
Allamandola et al. 1989);
in this case, we shall call the fluorescence process
described above "electronic fluorescence".
In addition, for highly isolated molecules, there could exist another two radiative energy decay channels - the inverse fluorescence (Leach 1987) and the recurrent fluorescence (Léger, Boissel, & d'Hendecourt 1988). The former results from a transition from a high vibrational level of a lower electronic state to a higher electronic state. The latter, also known as the Poincaré fluorescence, was postulated by Léger et al. (1988) as resulting from an inverse electronic conversion (i.e., the partial conversion of the vibrational energy of the ground electronic state into electronic excitation), followed by emitting a visible photon just like the ordinary (electronic) fluorescence process. However, the Poincaré fluorescence may have a quantum yield larger than one (i.e., several fluorescence photons can be emitted during one photoabsorption event since the molecule can oscillate many times between the electronic ground state and the excited state provided the absorbed energy is high enough; see Léger et al. 1988).
Figures 1 and 2 give a schematic overview of the above-described main processes following the absorption of an energetic photon respectively for a neutral molecule and a molecular cation.
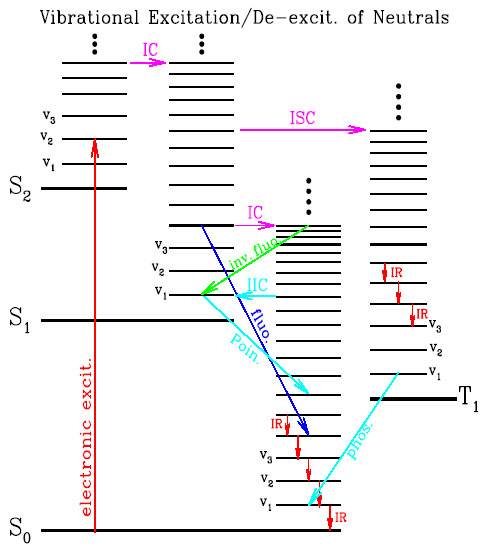 |
Figure 1. Step ladder model of a large neutral molecule. The schematic energy levels involve an electronic part (singlets S0, S1, S2 ... and triplets T1, T2 ...) and a vibrational part (v1, v2, v3...). Upon absorption of an UV/visible photon, the molecule makes an electronic transition from the ground state S0 to an upper state Sn, followed by (1) radiationless internal vibrational redistribution, (2) radiationless electronic transitions (internal conversion [IC] and intersystem crossing [ISC]), (3) radiative electronic transitions (fluorescence and phosphorescence), (4) radiative inverse electronic transitions (inverse fluorescence, and Poincaré fluorescence resulted from the inverse internal conversion [IIC]), and (5) IR vibrational transitions. |
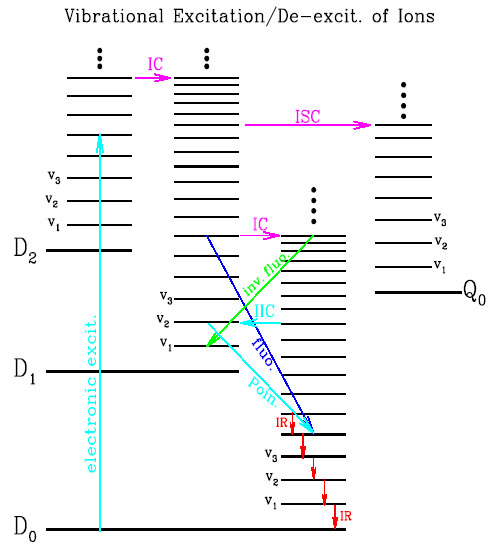 |
Figure 2. Same as Figure 1 but for a large molecular cation (with one unpaired electron). The schematic energy levels involve an electronic part (doublets D0, D1, D2 ... and quartets Q0, Q1, Q2 ...) and a vibrational part (v1, v2, v3...). In comparison with neutral molecules, radiative electronic transitions (fluorescence and phosphorescence) for ions are not important since internal conversion quickly transfers almost all of the initial excitation energy to the vibrationally excited ground electronic state D0 (see Leach 1987; Allamandola et al. 1989). Therefore, IR emission becomes almost the only deactivation route. |
The stochastic heating of ultrasmall grains has been extensively studied in literature (see Draine & Li 2001 and references therein). All studies prior to the identification of PAH molecules as the carrier of the "UIR" bands used the "thermal" approach (i.e., the vibrationally excited grain is described as a system having an internal temperature; see Section 3.3). The issue of "thermal" versus "non-thermal" arose when Léger and his coworkers used the "thermal" approach to model the vibrational excitation of PAHs to explain the "UIR" bands (Léger & Puget 1984; Léger, d'Hendecourt, & Défourneau 1989), while Allamandola, Tielens, & Barker (1985, 1989) took a statistical approach and questioned the validity of the "thermal" approximation. Barker & Cherchneff (1989), d'Hendecourt et al. (1989), Schutte, Tielens, & Allamandola (1993), and Cook & Saykally (1998) found that the thermal approximation was valid for computing thermal emission spectra. Below we will discuss the recent efforts we took to model the PAH excitation and de-excitation processes (Draine & Li 2001) using both the "exact-statistical" approach (Section 3.2) and the "thermal-approximation" approach (Section 3.3).